Molecular geometry and measurements¶
Geometry is MolScope's core. Every quantity below is computed in readable NumPy
and is cross-checked against MDAnalysis in the
validation suite (tests/validation/test_geometry_ref.py). A runnable tour of
all of them lives in examples/geometry.py.
import molscope as ms
mol = ms.read("examples/data/1fqy.pdb")
Distances, angles, dihedrals¶
The three local internal coordinates, all in one structure:
mol.distance(i, j) # |r_i - r_j|, in angstrom
mol.angle(i, j, k) # angle at j, in degrees
mol.dihedral(a, b, c, d) # torsion about the b-c bond, in degrees (-180, 180]
- Distance is the Euclidean norm of the difference vector.
- Angle at the central atom
jcomes from the dot product of the two bond vectors:acos((u·v)/(|u||v|)). - Dihedral is the angle between the two planes sharing the
b-cbond, computed with the numerically stableatan2formulation, so it carries the correct sign (the basis of backbone phi/psi torsions).
Centroid vs centre of mass¶
Two notions of "the middle", and the difference matters:
mol.centroid # unweighted mean of positions
mol.center_of_mass # mass-weighted: sum(m_i r_i) / sum(m_i)
The centroid treats every atom equally; the centre of mass pulls towards
heavy atoms. They coincide for a homonuclear system and drift apart when heavy
atoms sit off-centre. Centre on either with mol.centered() (centroid) or
mol.centered(weighted=True) (COM).
Radius of gyration¶
A single number for overall size and compactness:
mol.radius_of_gyration # Rg, in angstrom
Rg = sqrt(sum(m_i |r_i - R_com|^2) / sum(m_i)) is the mass-weighted RMS
distance of atoms from the centre of mass. It is small for globular structures
and grows as a chain extends or unfolds, which makes it a handy reaction
coordinate for folding and compaction.
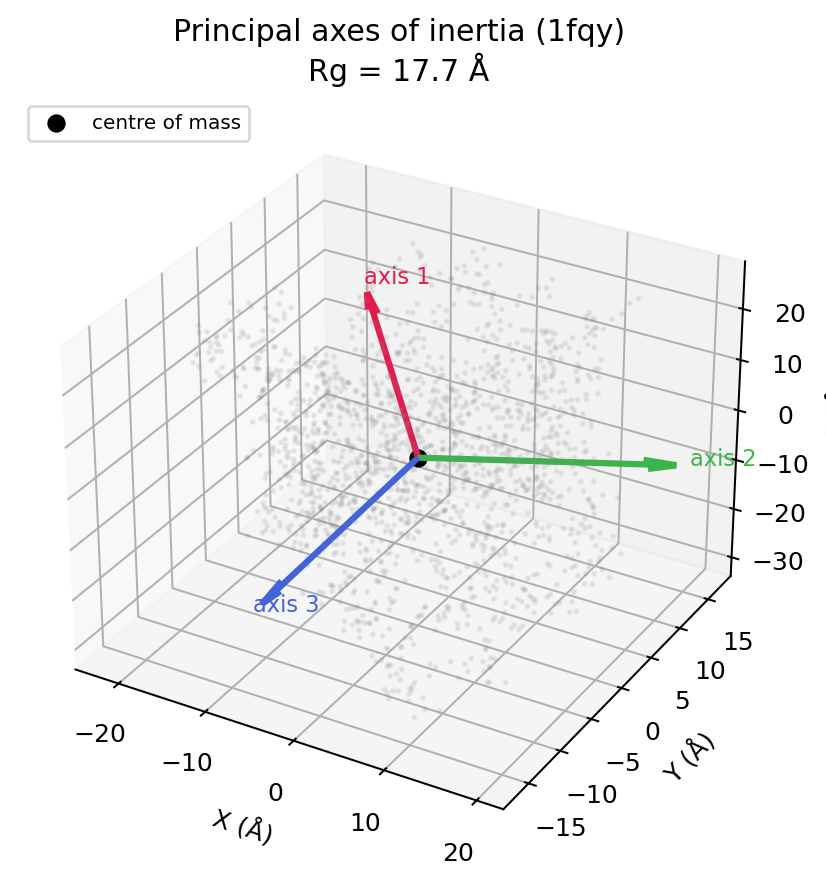
Inertia tensor and principal axes¶
How mass is distributed in space, i.e. the molecule's overall shape:
mol.inertia_tensor() # (3, 3) mass-weighted tensor about the COM
mol.principal_moments() # (3,) eigenvalues, ascending
mol.principal_axes() # (3, 3) eigenvectors as columns, moment-sorted
The inertia tensor is I = sum_i m_i (|r_i|^2 I_3 - r_i r_i^T) about the centre
of mass. Diagonalising it gives the principal axes (the natural body frame)
and the principal moments (how mass spreads along each axis). Equal moments
mean a spherical mass distribution; one small and two large moments mean a rod;
the ratios drive shape descriptors like asphericity.
Kabsch alignment and RMSD¶
To compare two structures you must first remove rigid-body differences. The Kabsch algorithm finds the rotation (via an SVD of the cross-covariance, with a reflection correction) that best superposes one structure onto another:
aligned = a.superpose(b) # a optimally rotated/translated onto b
rmsd = a.rmsd(b, align=True) # RMSD after that optimal fit
raw = a.rmsd(b) # RMSD as-is, no alignment
RMSD (root-mean-square deviation) is sqrt(mean(|a_i - b_i|^2)) over matched
atoms. With align=True it is the minimum RMSD over all rigid orientations, the
standard measure of how similar two conformations are.
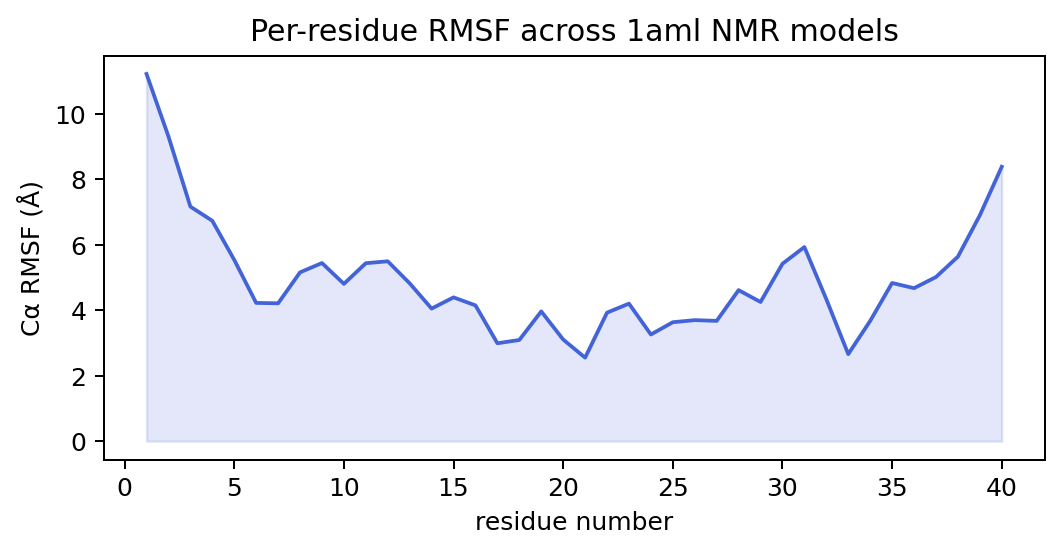
Ensembles: RMSF and the RMSD matrix¶
Across a set of structures (an NMR ensemble, conformers, a trajectory), MolScope summarises motion and spread:
models = ms.read_pdb_models("examples/data/1aml.pdb")
ms.ensemble.rmsf(models) # per-atom root-mean-square fluctuation
ms.rmsd_matrix(models) # (M, M) pairwise RMSD between models
ms.ensemble.average(models) # mean structure
RMSF (root-mean-square fluctuation) measures how much each atom moves about its mean position after alignment, so it maps flexibility onto the sequence: flexible loops and termini spike, the structured core stays low.
The pairwise RMSD matrix shows how the models relate to each other and feeds
conformer clustering (ms.cluster); plot it with ms.plot_rmsd_heatmap.
Pairwise distances and contacts¶
D = mol.distance_matrix() # dense (N, N), NumPy by default
pairs = mol.contacts(cutoff=5.0) # atom index pairs within a cutoff
distance_matrix() supports NumPy, PyTorch, CuPy, and auto backends; see
Contact maps and distance matrices for backends, images, and
benchmarks.