Coarse-Grain A Protein¶
This example is for learning and inspection. It does not create production simulation topologies or validated Martini parameters.
import molscope as ms
mol = ms.read("examples/data/1fqy.pdb")
cg = mol.coarse_grain("residue_com")
print(cg.summary())
print(cg.mapping_report())
cg.plot(scale=200)
For a Martini-like teaching model:
cg = mol.coarse_grain("martini")
G = cg.to_graph()
Conceptually, Martini-style coarse-graining groups atoms into interaction sites
such as backbone and sidechain beads. MolScope's "martini" mode only teaches
that mapping idea: it creates BB/SC bead coordinates and a simple bead graph.
Real Martini force fields additionally require validated bead types, bonded and
nonbonded parameters, charges, exclusions, and simulation-engine topology files.
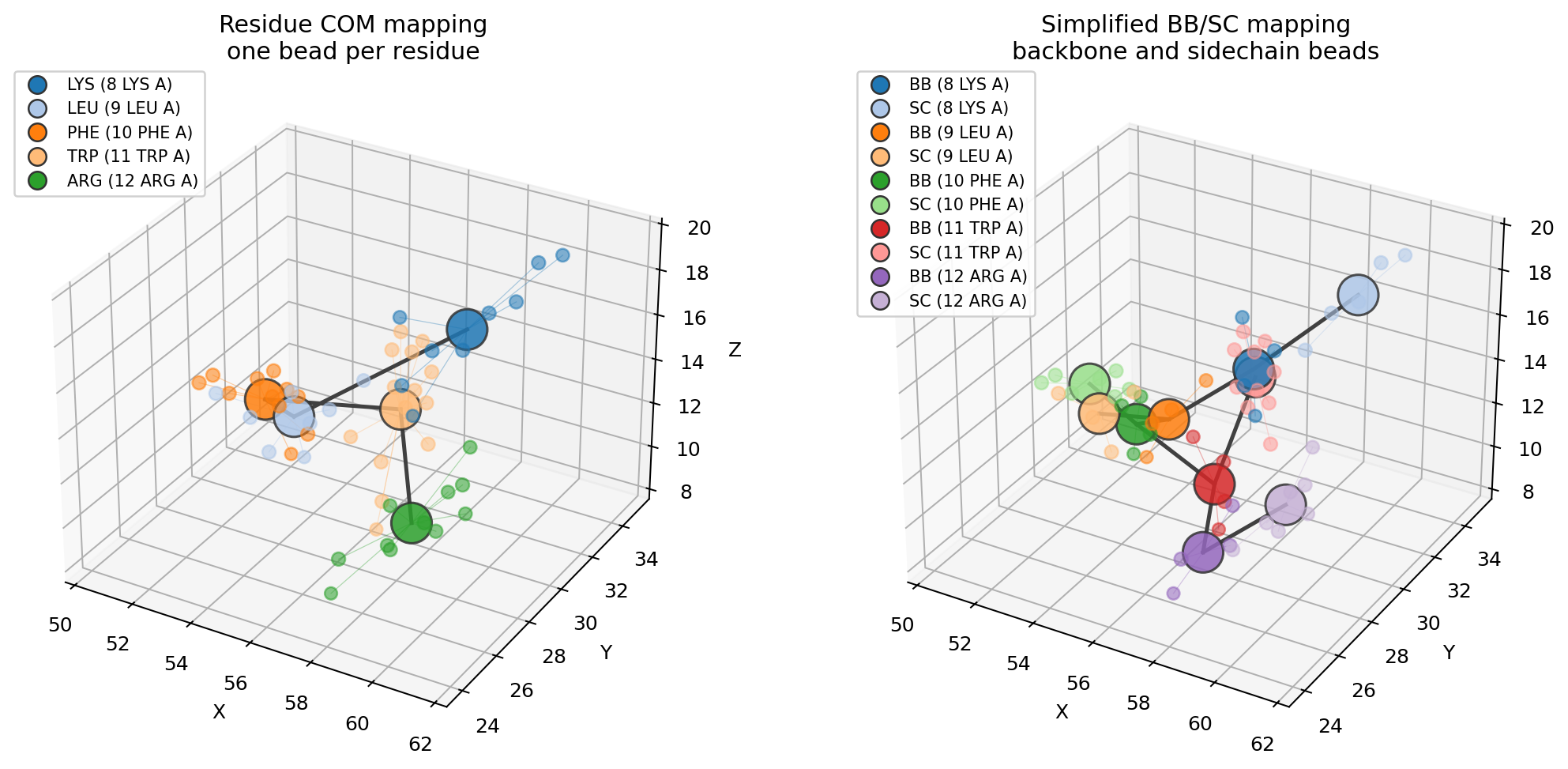
See the mapping¶
plot_mapping overlays the beads on the atoms they replace, colouring each atom
by its bead. A short fragment reads most clearly:
fragment = mol.select(resid=(8, 12))
ms.plot_mapping(fragment, fragment.coarse_grain("martini"))
For a side-by-side residue COM vs backbone/sidechain visual:
uv run python examples/coarse_graining.py
Inspect and export the assignment¶
report = cg.coarse_grain_report
print(report.coverage()) # beads / atoms covered
print(report.beads[0].atom_indices) # which atoms became bead 0
cg.write_mapping("mapping.json") # JSON record (round-trippable)
cg.write_index("mapping.ndx") # GROMACS-style index, one group per bead
record = ms.read_cg_mapping("mapping.json")
cg_again = ms.apply_cg_mapping(mol, record) # rebuild on the same structure