Contact maps and distance matrices¶
Contact maps and dense distance matrices are MolScope's most direct structural analysis tools. They are useful for checking folds, comparing ensembles, building ML features, and validating coarse-grained mappings.
Dense distance matrices¶
import molscope as ms
mol = ms.read("examples/data/1fqy.pdb")
ca = mol.alpha_carbons()
D = ca.distance_matrix() # NumPy CPU backend
print(D.shape) # (226, 226)
ca.plot_distance_matrix(show=False)
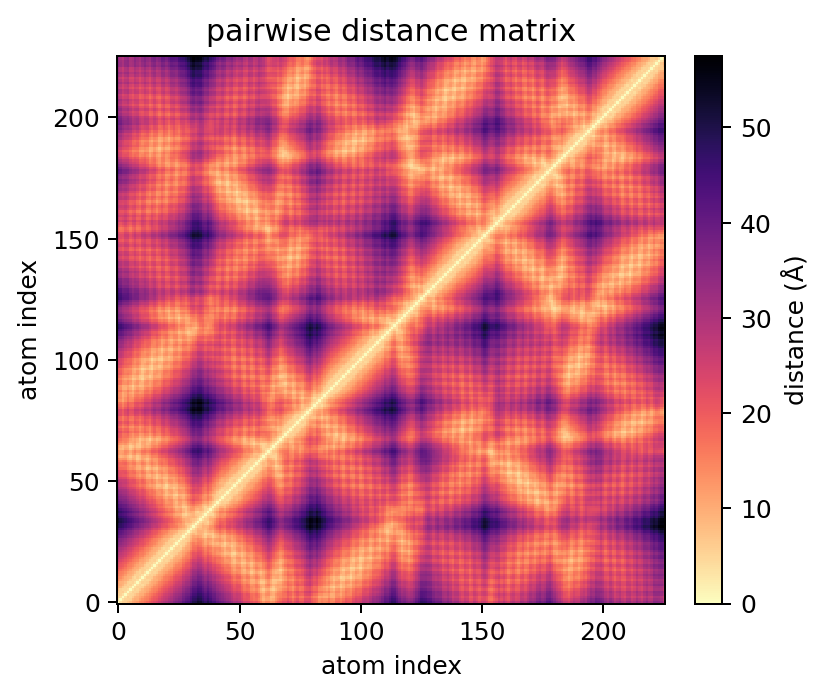
By default, distance_matrix() returns a NumPy array. Optional dense backends
can be selected when you already have the corresponding array stack installed:
D = ca.distance_matrix(backend="numpy") # CPU NumPy
D = ca.distance_matrix(backend="torch", device="cpu") # PyTorch CPU
D = ca.distance_matrix(backend="torch", device="cuda") # PyTorch CUDA
D = ca.distance_matrix(backend="cupy") # CuPy CUDA
D = ca.distance_matrix(backend="auto") # GPU if available
Results are converted back to NumPy by default so plotting and downstream MolScope APIs keep working. Advanced users can keep a backend-native array:
torch_matrix = ca.distance_matrix(backend="torch", device="cuda", as_numpy=False)
Atom contacts¶
contacts() returns atom index pairs within a cutoff:
pairs = mol.contacts(cutoff=5.0)
count = mol.contact_count(cutoff=5.0)
The default contacts() path uses SciPy's KD-tree when SciPy is installed and a
chunked NumPy fallback otherwise. Dense backends are also available:
pairs = mol.contacts(cutoff=5.0, backend="numpy")
pairs = mol.contacts(cutoff=5.0, backend="torch", device="cuda")
Use dense backends when you already need a full matrix or want GPU acceleration for moderate-sized structures. For sparse pair lists on large structures, the KD-tree path is usually the better CPU choice.
Residue contact maps¶
Residue contact maps summarize which residue pairs are closer than a cutoff:
cmap = mol.contact_map(cutoff=8.0, level="residue", method="ca")
cmap.matrix
cmap.labels[:3]
cmap.plot(show=False)
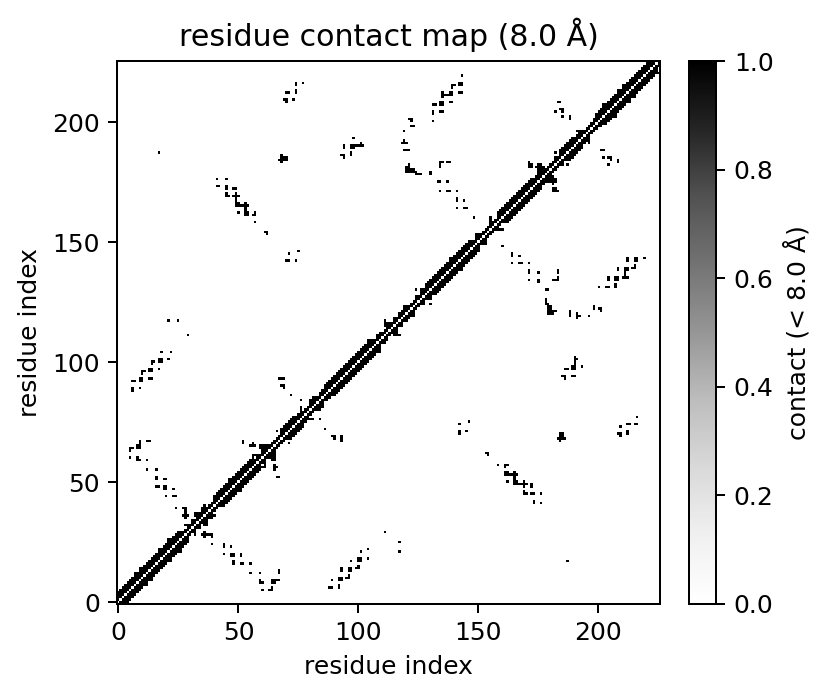
Residue methods:
method="ca": alpha-carbon distance, with centroid fallback when CA is absent.method="com": residue centre-of-mass distance.method="min": closest inter-residue atom pair.
Dense backends apply to residue representative distances and atom-level contact maps:
cmap = mol.contact_map(cutoff=8.0, level="residue", backend="torch", device="cuda")
atom_map = mol.contact_map(cutoff=5.0, level="atom", backend="cupy")
Filtering by sequence separation and chain¶
Two filters narrow a map to the contacts you care about:
# Drop trivial local contacts (i, i+1 ... i+3 within a chain).
longrange = mol.contact_map(cutoff=8.0, level="residue", min_seq_sep=4)
# Keep only intra-chain or only inter-chain pairs (multi-chain structures).
intra = mol.contact_map(cutoff=8.0, level="residue", chain_mode="intra")
inter = mol.contact_map(cutoff=8.0, level="residue", chain_mode="inter")
min_seq_sep removes same-chain pairs closer than that many positions in
sequence; chain_mode is "all" (default), "intra", or "inter".
Contact metrics¶
cmap = mol.contact_map(cutoff=8.0, level="residue", min_seq_sep=4)
cmap.n_contacts # number of contacting residue pairs
cmap.contact_order() # relative contact order (local vs long-range)
Relative contact order is the mean sequence separation of contacts divided by chain length: low for helical/local folds, higher when many long-range contacts are present. It is most meaningful for a single chain.
Labelled heatmaps¶
For small maps, plot() annotates the axes with residue labels (e.g.
A:LYS8); force this on or off with label_ticks:
peptide.contact_map(level="residue").plot(label_ticks=True, show=False)
Ensemble contact frequency¶
For NMR ensembles or conformer sets, contact frequency reports how often each pair is in contact:
models = ms.read_pdb_models("examples/data/1aml.pdb")
freq = ms.ensemble_contact_frequency(models, cutoff=8.0)
freq.plot(show=False)
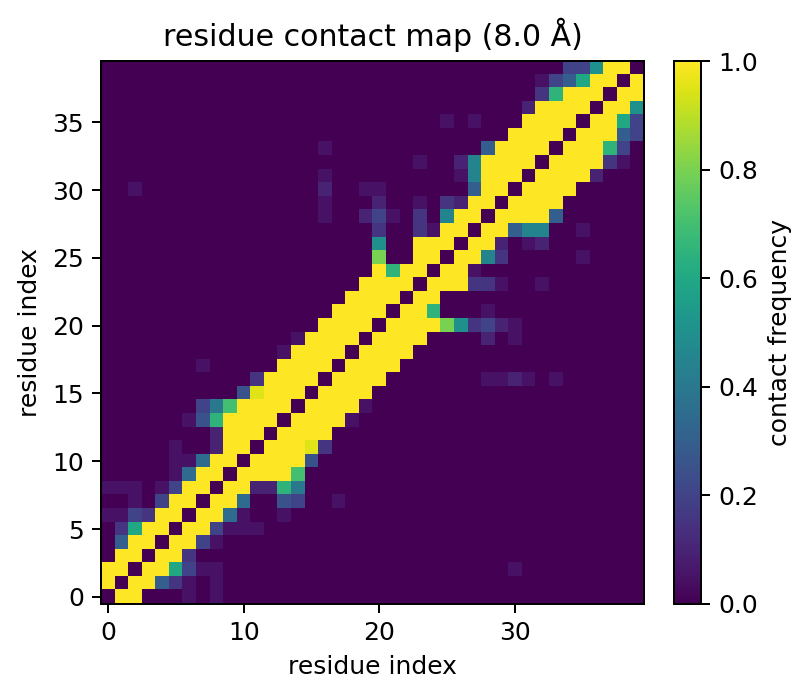
The matrix values are in [0, 1], where 1.0 means a pair is in contact in
every model and intermediate values indicate conformational variability.
Backend guidance¶
| Backend | Use when | Notes |
|---|---|---|
numpy |
You want predictable CPU dense matrices | Always available |
scipy |
You want sparse atom contact pairs/counts on CPU | Default for contacts() when installed |
torch |
You already use PyTorch or want CUDA/MPS dense matrices | Install platform-specific PyTorch first |
cupy |
You already use CUDA CuPy arrays | Install the CUDA-matched CuPy package |
auto |
You want MolScope to pick GPU if available | Falls back to NumPy |
Dense matrices are O(N^2) memory. Prefer residue maps, alpha-carbon subsets,
or KD-tree contact pairs for large atomistic systems.